|
|
|
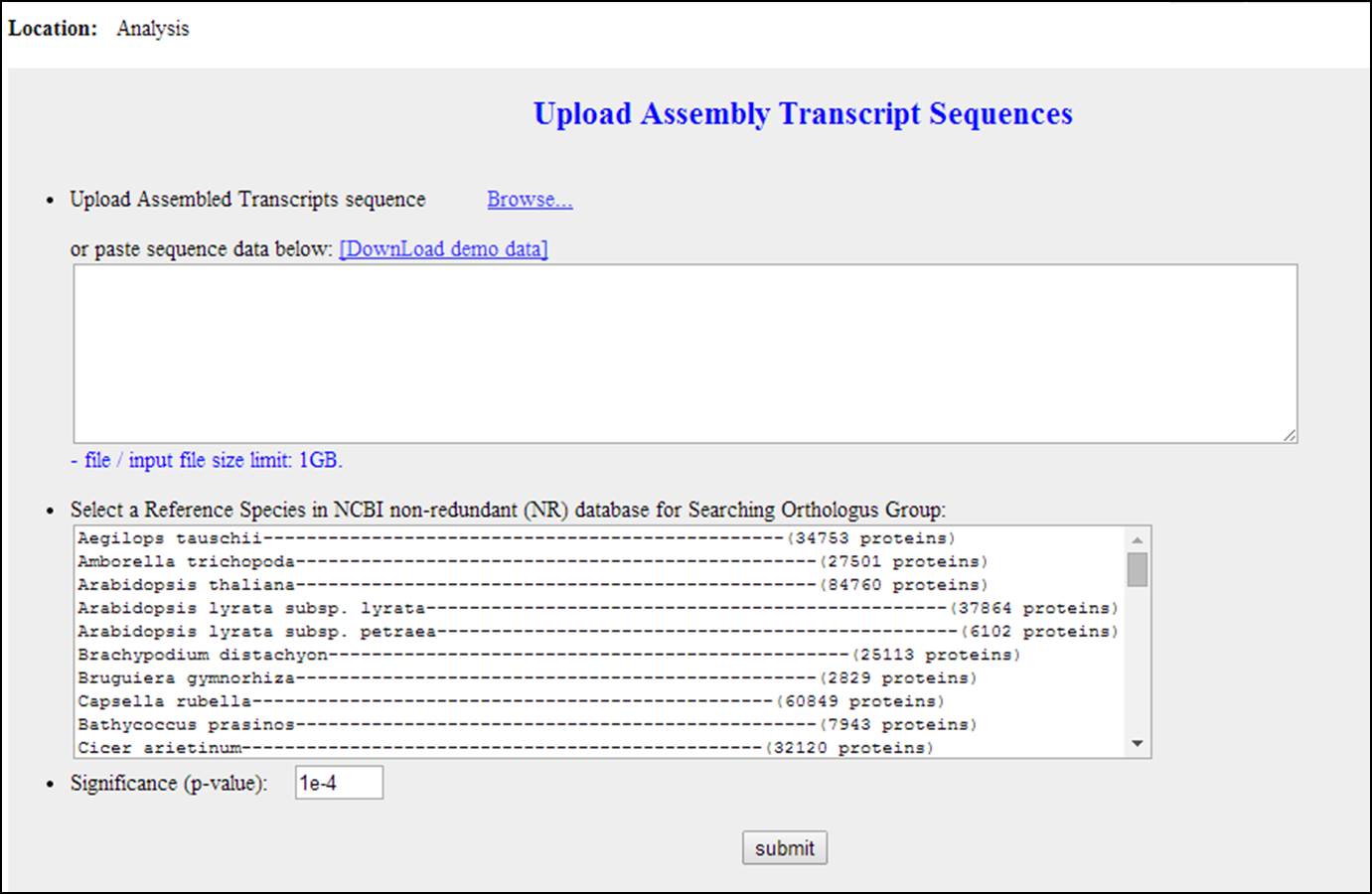
Input for sequence annotation based on NCBI NR database
The input file should be a sequence file in a fasta format;
|
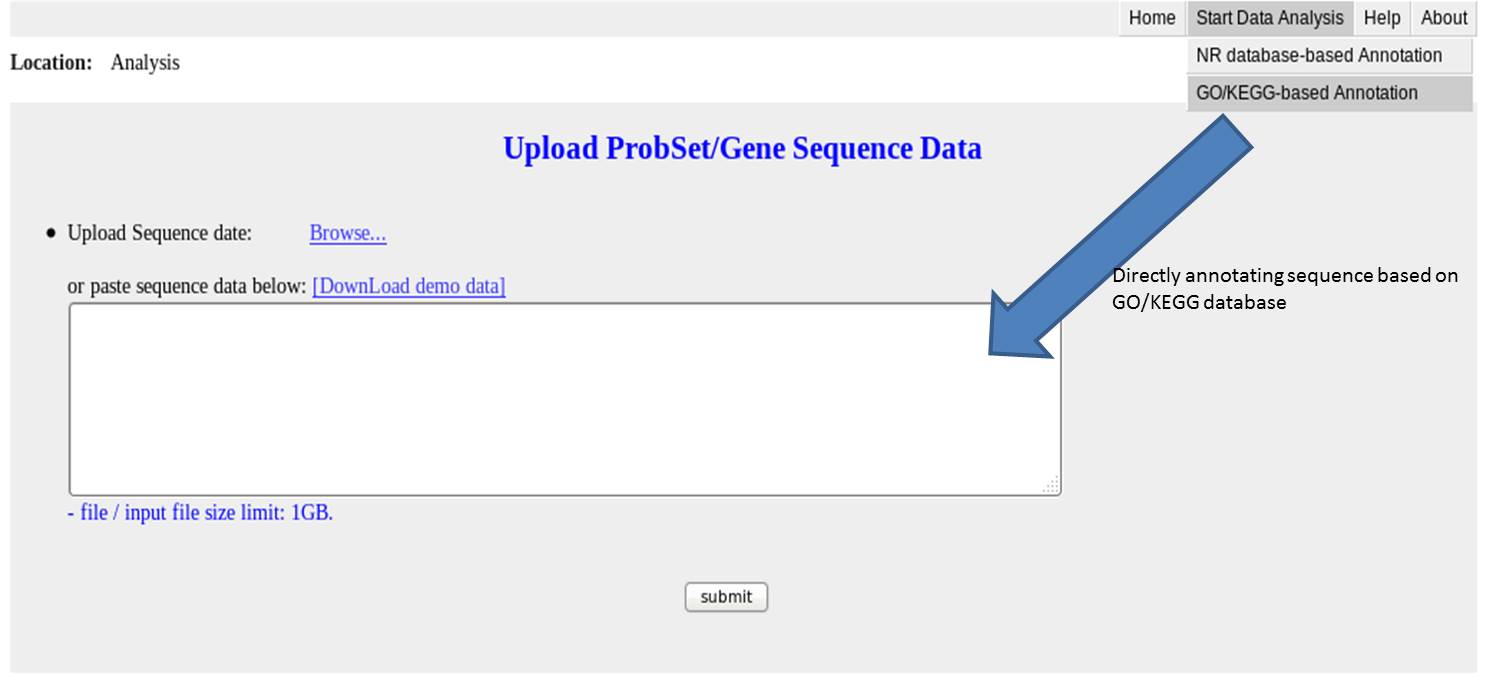
Input for sequence annotation based on GO/KEGG database without using NCBI NR database as a proxy
The input file should be a sequence file in a fasta format;
|
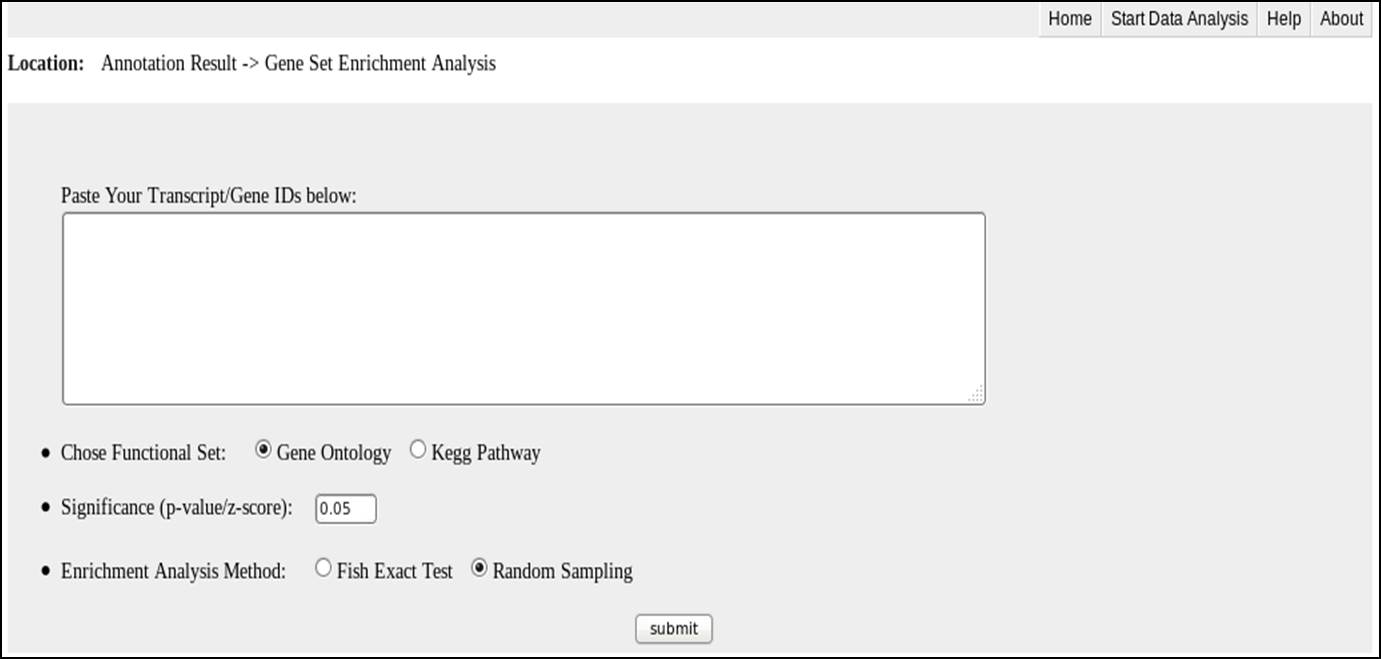
Input for Transcript/Gene Enrichment analysis
The input file is user defined Transcript/Gene list:: please only input one Transcript/Gene ID per line;
|
|
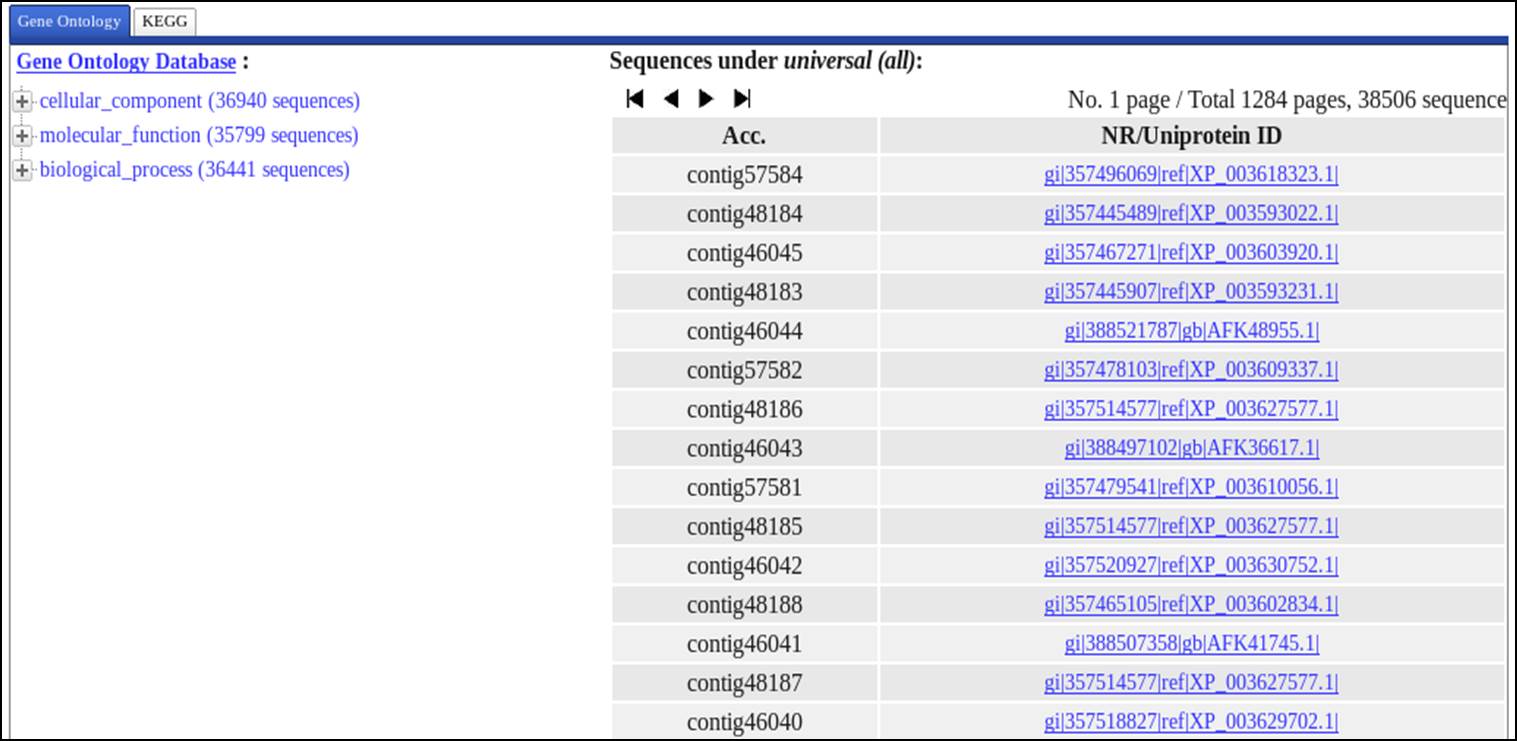
Output1
Annotation Result based on Gene Ontology Analysis
|
|
Output2
Enrichment Analysis Result based on Gene Ontology

|
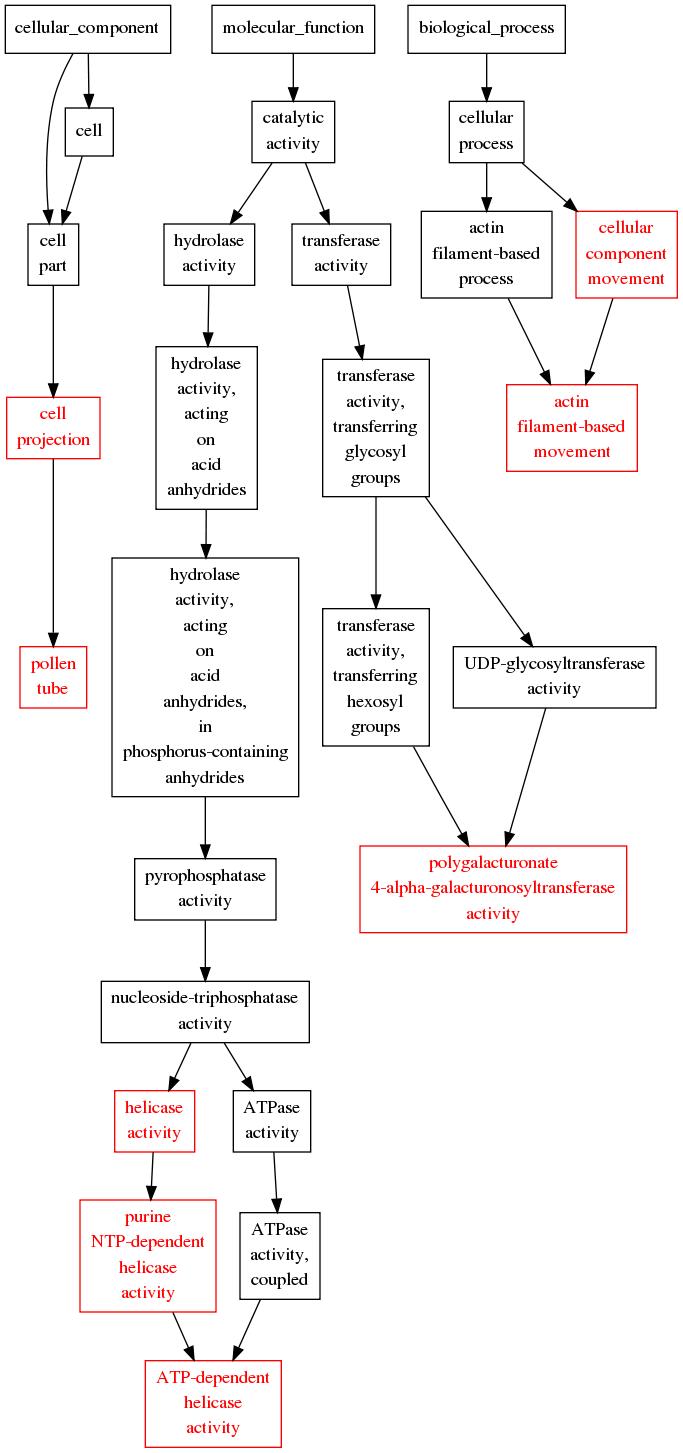
Visualize Enrichment Result with Graphviz
Funding:
- This work was supported by National Science Foundation (Grant ABI-0960897) and the Samuel Roberts Noble Foundation.
Contact:
- To contact us, please write to: bioinfo AT noble DOT org
|
| |
© 2013-2014 by The Samuel Roberts Noble Foundation, Inc. Site by Zhao Lab |
|
