GWASpro user manual
Data protection policy
Uploaded data and results are saved in a short-term unique session folder that expire in two weeks. All data in the session will be automatically deleted from our web server after expiration. Each session will receive a unique URL (ID) which are only available to the user who submit this job. User can only access their data and result by this URL. The unique URL is not visible to public and it is encrypted using HTTPS/SSL protocols. We do not look into these session data for any purposes other than counting service/hardware usages.
Please note:
- Genotypic code set can vary depending on users, so users can go with either (-1 for aa , 0 for Aa, 1 for AA,) or (0 for aa, 1 for Aa, 2 for AA); here, aa, Aa, AA corresponds to minor homogeneity, heterogeneity and major homogeneity.
- Input values are case-insensitive.
- Populations stratification columns in PCA or STRUCTURE tables must be numerical.
- Environmental factor colums (e.g. replications, treatments, locations, years) must be categorical.
- Input files can be uploaded from a user's computer, or the links of input files can be specified.
- Genomic data imputation must be made prior to submitting to GWASpro. A list of imputation tools can be found at https://omictools.com/genotype-imputation-category.
GWASpro screenshot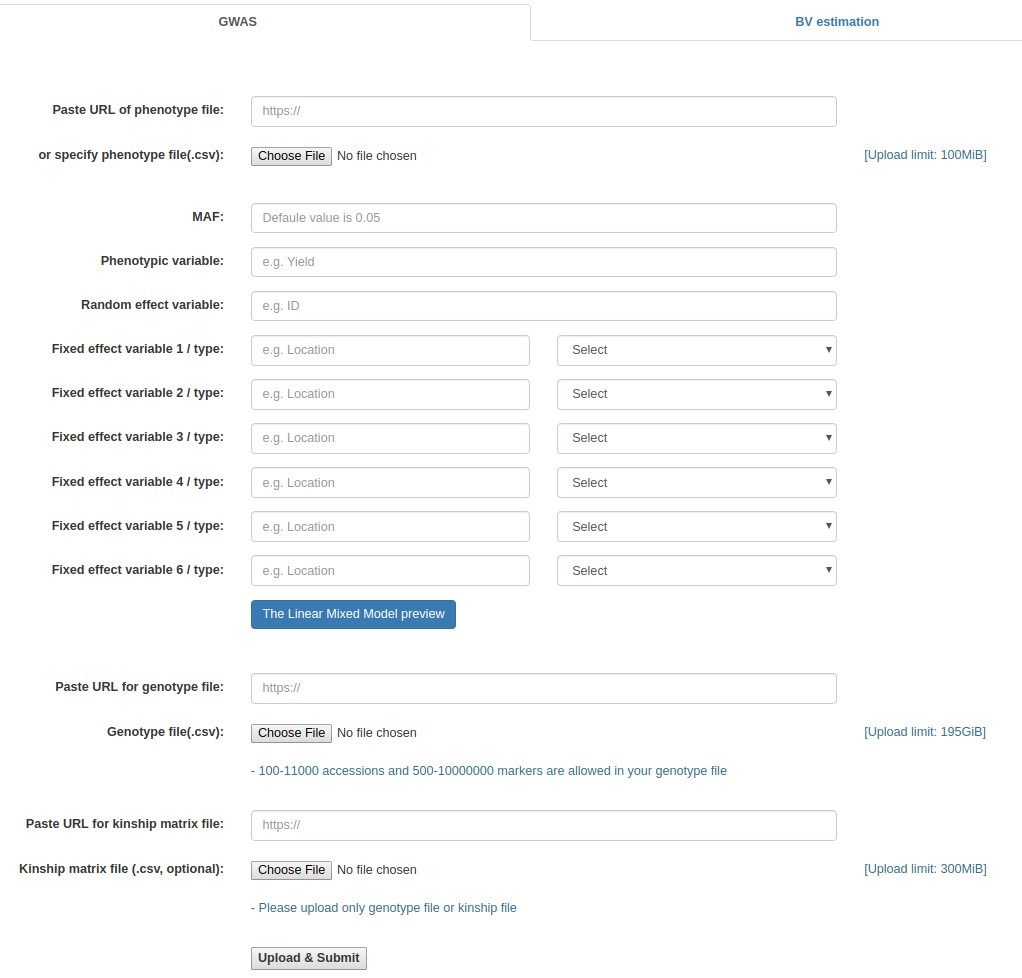
|
|---|
Case study 1: Rice data (Uploarding of input files)
- Download the following three files:
- Rice genotypic data (rice_genotype.csv)
- Rice phenotypic data (rice_phenotype.csv)
- Rice kinship matrix (rice_kinship.csv)
- Click the "Browse" button next to the "Phenotype file" field, go to the folder where the input data files are saved, and choose the following file: "rice_phenotype.csv."
- Fill out the "Phenotypic variable" field with either of "Yield", "KGW", "Tiller", and "Grain". These are column labels of traits in the "rice_phenotype.csv" file.
- Fill out the "Random effect variable" field with "Plant", which is the 2nd column label of the "rice_phenotype.csv" file.
- Fill out the "Fixed effect variable 1" field with "PC1", which is the 6th column label of the "rice_phenotype.csv" file. Subsequently, select "Numerical".
- Fill out the "Fixed effect variable 2" field with "PC2", which is the 7th column label of the "rice_phenotype.csv" file. Subsequently, select "Numerical".
- Fill out the "Fixed effect variable 3" field with "PC3", which is the 8th column label of the "rice_phenotype.csv" file. Subsequently, select "Numerical".
- Click the "Browse" button for "Genotype file", and choose the following file: "rice_genotype.csv".
- Click the "Browse" button for "Kinship matrix file" field, and choose the following file: "rice_kinship.csv". This file upload is optional. If not uploaded, the kinship matrix will be internally calculated based on the uploaded genotypic data set.
- Click the "Upload & Submit" button.
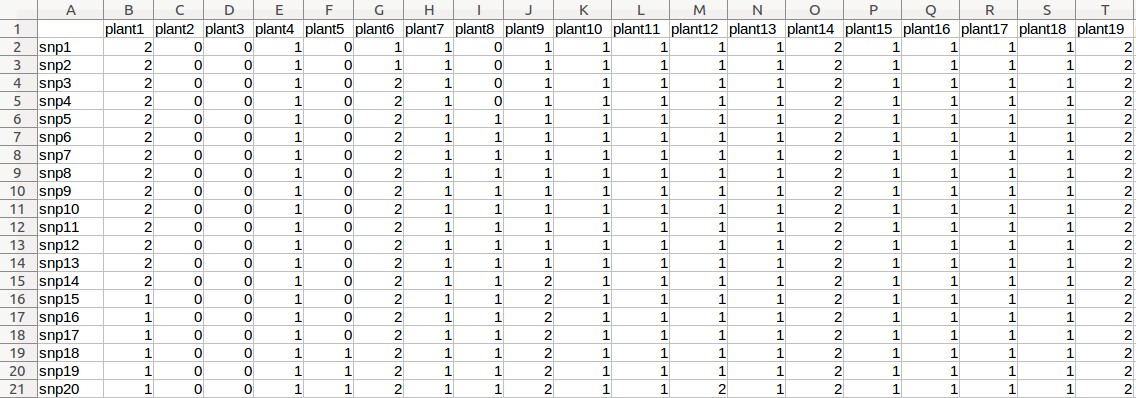
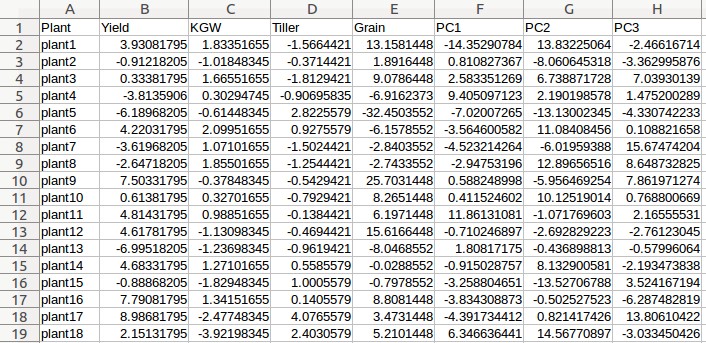
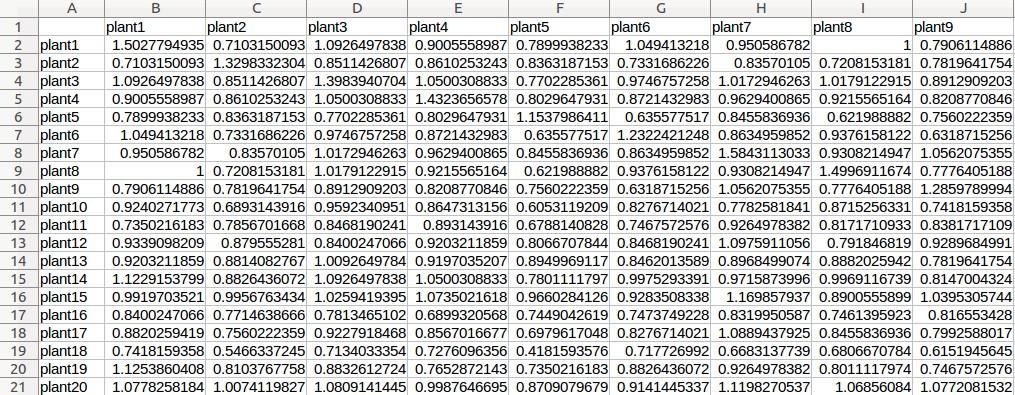
Example - Uploading of input files (Rice data)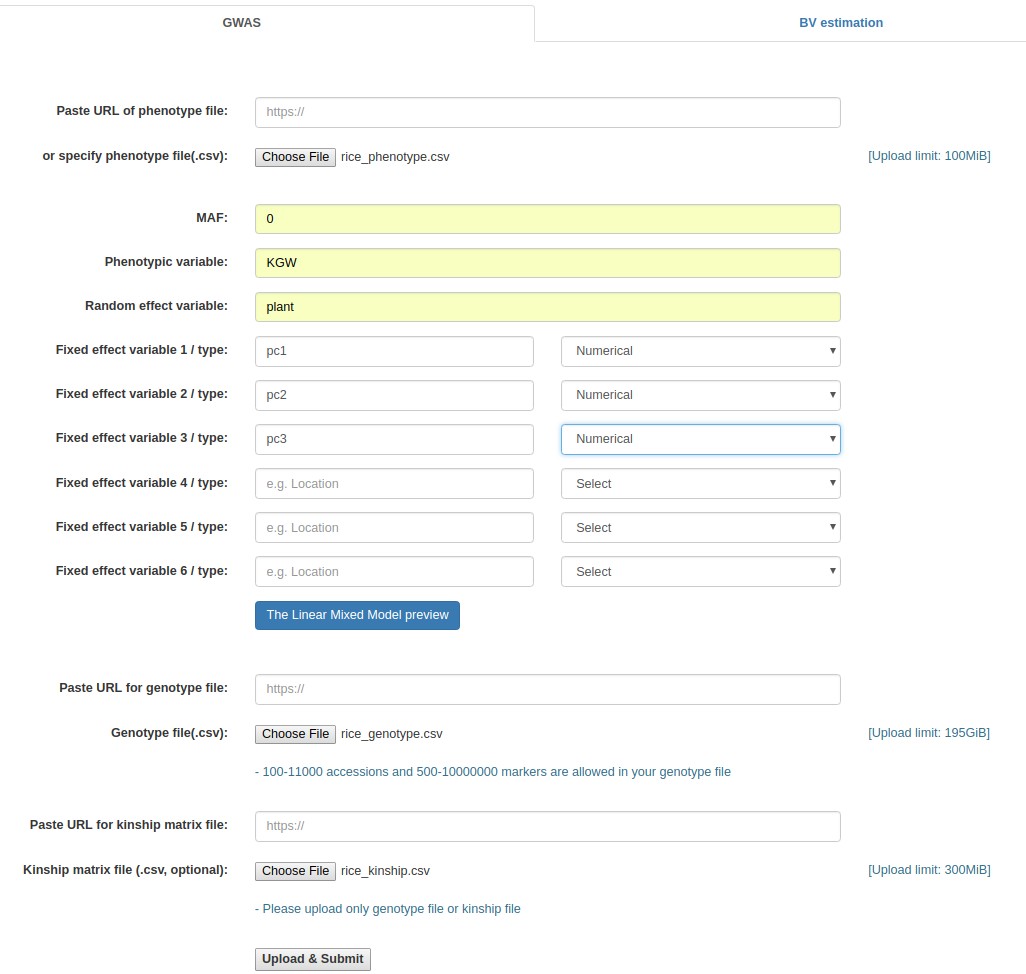
|
|---|
Case study 1: Rice data (Specifying the online paths of the input files)
- Fill out the "Paste URL of phenotype file" field with "http://www.zhaolab.org/GWASPRO/static/demo/rice_phenotype.csv".
- Fill out the "Phenotypic variable" field with either of "Yield", "KGW", "Tiller", and "Grain". These are column labels of traits in the "rice_phenotype.csv" file.
- Fill out the "Random effect variable" field with "Plant", which is the 2nd column label of the "rice_phenotype.csv" file.
- Fill out the "Fixed effect variable 1" field with "PC1", which is the 6th column label of the "rice_phenotype.csv" file. Subsequently, select "Numerical".
- Fill out the "Fixed effect variable 2" field with "PC2", which is the 7th column label of the "rice_phenotype.csv" file. Subsequently, select "Numerical".
- Fill out the "Fixed effect variable 3" field with "PC3", which is the 8th column label of the "rice_phenotype.csv" file. Subsequently, select "Numerical".
- Fill out the "Paste URL for genotype file" field with "http://www.zhaolab.org/GWASPRO/static/demo/rice_genotype.csv".
- Fill out the "Paste URL for kinship matrix file" field with "http://www.zhaolab.org/GWASPRO/static/demo/rice_kinship.csv".
- Click the "Upload & Submit" button.
Example - Specifying the online file paths (Rice data)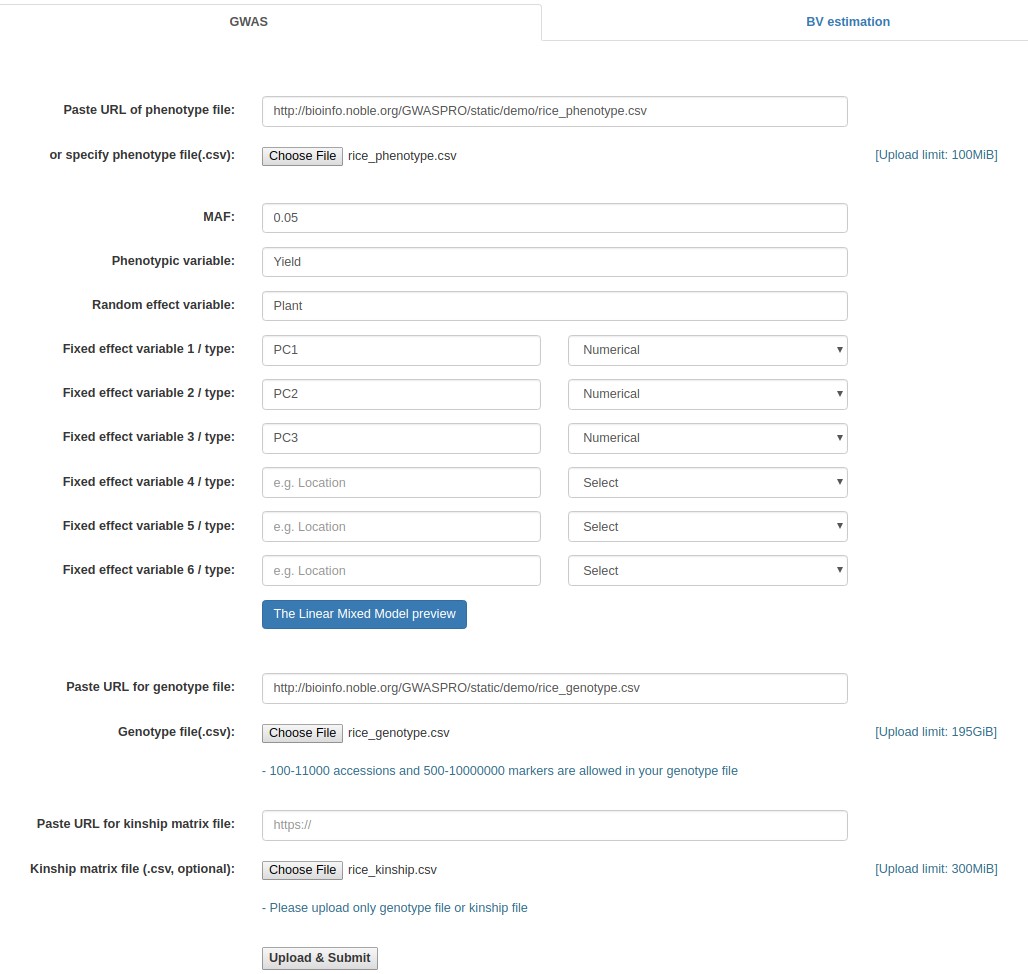
|
|---|
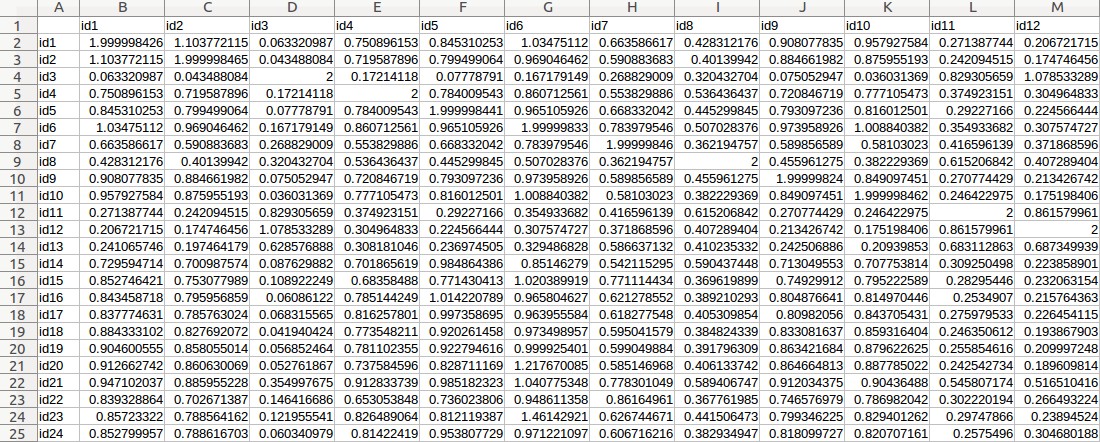
Case study 2: Medicago truncatula data (Uploarding of input files)
- Download the following four files:
- Medicago genotypic data (Medicago_geno.csv)
- Medicago phenotypic data (Medicago_pheno.csv)
- Medicago kinship matrix (Medicago _kinship.csv)
- SNP_map (SNP_map_medicago.csv)
- Click the "Browse" button next to the "Phenotype file" field, go to the folder where the input data files are saved, and choose the following file: "Medicago_pheno.csv."
- Fill out the "MAF" field with 0.
- Fill out the "Phenotypic variable" field with either "LeafSize" or "shootDryWeight". These are column labels of traits in the "Medicago _pheno.csv" file.
- Fill out the "Random effect variable" field with "id", which is the 1st column label of the "Medicago _pheno.csv" file.
- Fill out the "Fixed effect variable 1" field with "Q1", which is the 5th column label of the "Medicago _pheno.csv" file. Subsequently, select "Numerical".
- Click the "Browse" button for "Genotype file", and choose the following file: "Medicago _geno.csv".
- Click the "Browse" button for "Kinship matrix file", and choose the following file: "Medicago_Kinship.csv". This file upload is optional.
- Click the "Upload & Submit" button.
- Once the analysis is completed, you can download P value result file. If you want to have GWASpro generating a Manhattan plot, click "manhattan plot" link, which will direct you to "MPlot – Manhattan Plot Draw" page.
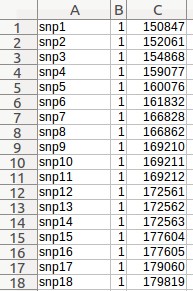
- Click the button next to "Marker Coordinates file" field, and choose the "SNP_map_medicago.csv" file.
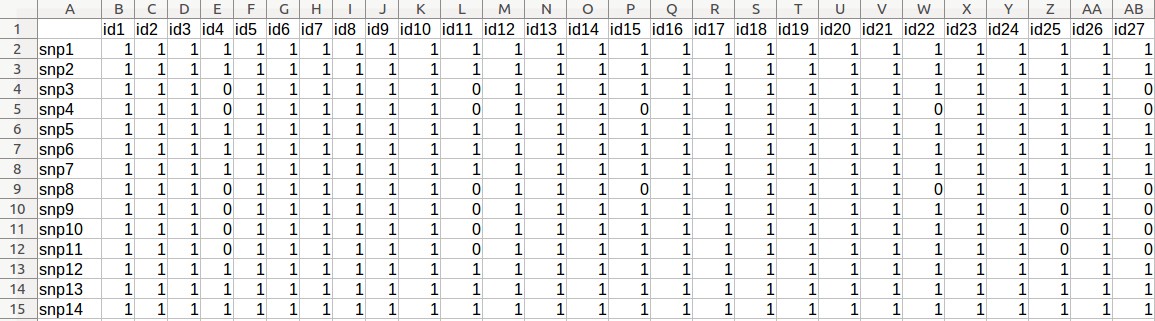
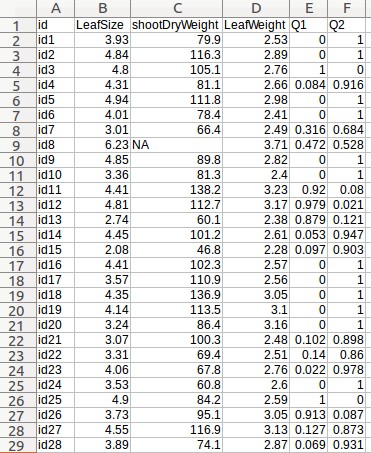
Example - Uploading of input files (Medicago truncatula data)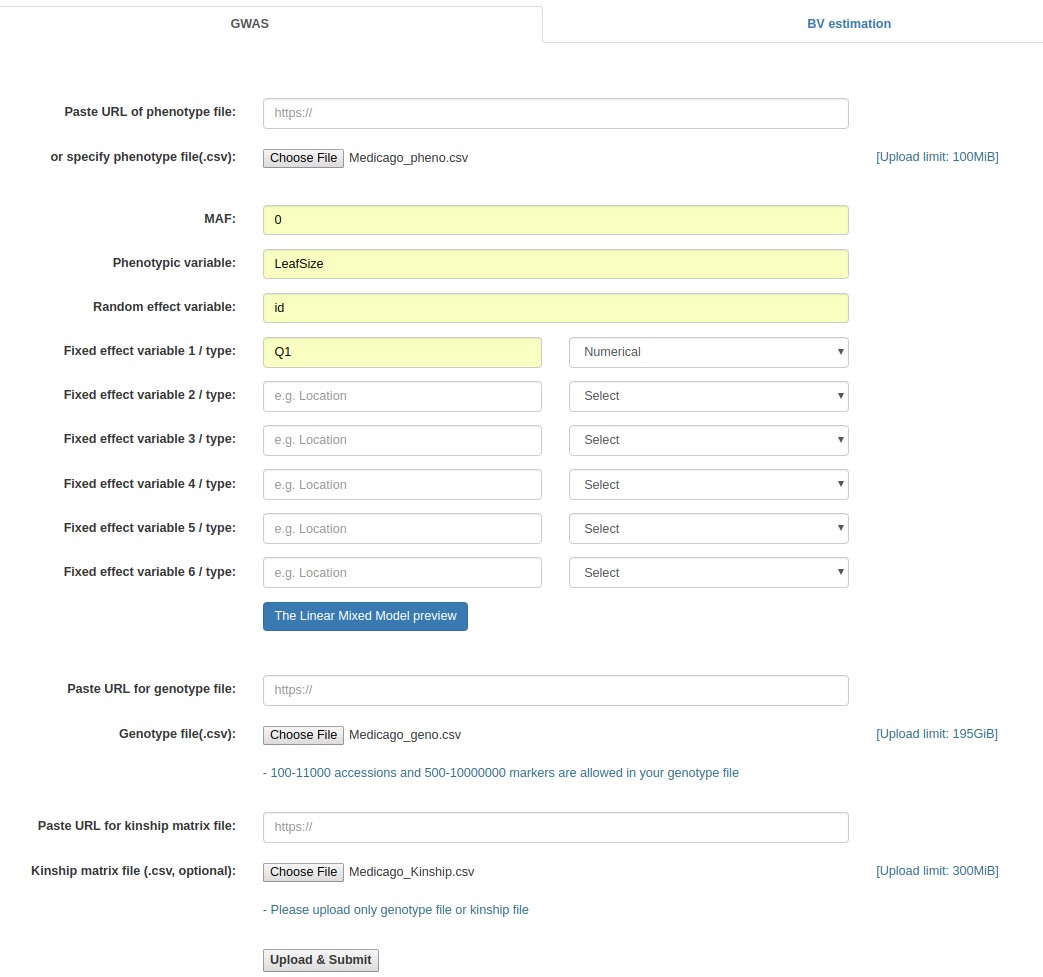
|
|---|
Case study 2: Medicago truncatula data (Specifying the online paths of the input files)
- Fill out the "Paste URL of phenotype file" field with "http://www.zhaolab.org/GWASPRO/static/demo/Medicago_pheno.csv".
- Fill out the "MAF" field with 0.
- Fill out the "Phenotypic variable" field with either "LeafSize" or "shootDryWeight". These are column labels of traits in the "Medicago _pheno.csv" file.
- Fill out the "Random effect variable" field with "id", which is the 1st column label of the "Medicago _pheno.csv" file.
- Fill out the "Fixed effect variable 1" field with "Q1", which is the 5th column label of the "Medicago _pheno.csv" file. Subsequently, select "Numerical".
- Fill out the "Paste URL for genotype file" field with "http://www.zhaolab.org/GWASPRO/static/demo/Medicago_geno.csv".
- Fill out the "Paste URL for kinship matrix file" field with "http://www.zhaolab.org/GWASPRO/static/demo/Medicago_kinship.csv". This file is optional.
- Click the "Upload & Submit" button.
- Once the analysis is completed, you can download P value result file. If you want to have GWASpro generating a Manhattan plot, click "manhattan plot" link, which will direct you to "MPlot – Manhattan Plot Draw" page.
- Click the button next to "Marker Coordinates file" field, and choose the "SNP_map_medicago.csv" file.
Example - Specifying the online file paths (Medicago truncatula data)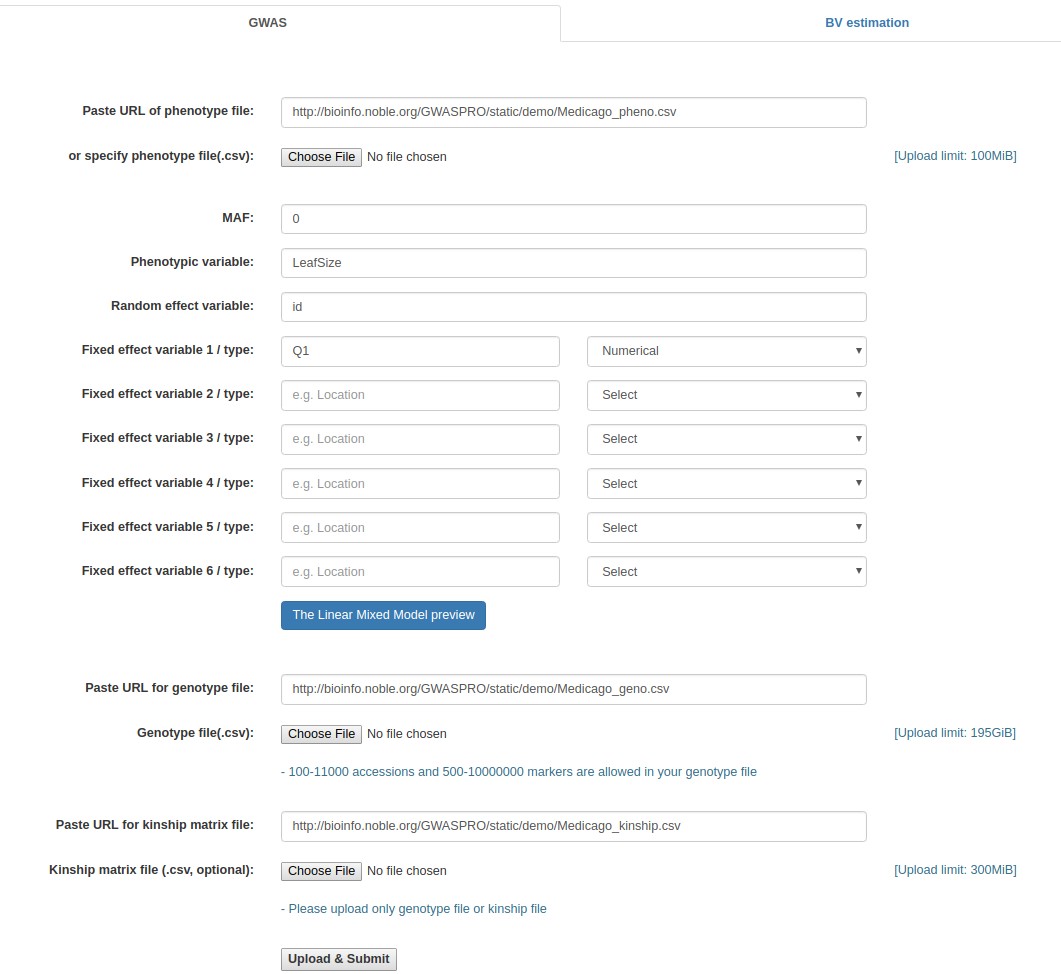
|
|---|
Case study 3: Maize data (Uploarding of input files)
- Download the following three files:
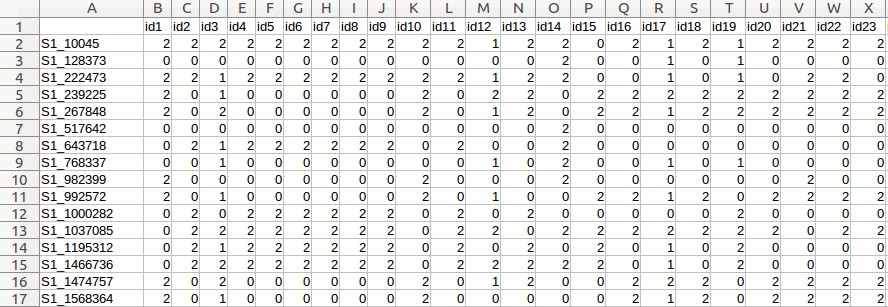
- Maize genotypic data (Maize_geno.csv)
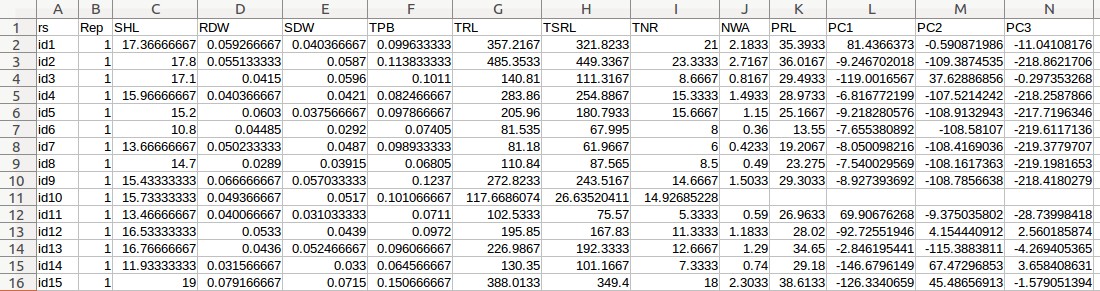
- Maize phenotypic data (Maize_pheno_pca.csv)
- SNP_map (SNP_map_maize.csv)
- Click the "Browse" button next to the "Phenotype file" field, go to the folder where the data files are saved, and choose the following file: "Maize_pheno_pca.csv."
- Fill out the "MAF" field with 0.
- Fill out the "Phenotypic variable" field with either "TNR" or "TRL". These are column labels of traits in the "Maize _pheno_pca.csv" file.
- Fill out the "Random effect variable" field with "rs", which is the 1st column label of the "Maize _pheno.csv" file.
- Fill out the "Fixed effect variable 1" field with "Rep", which is the 2nd column label of the "Maize _pheno.csv" file. Subsequently, select "Categorical".
- Fill out the "Fixed effect variable 2" field with "PC1", which is the 12th column label of the "Maize _pheno.csv" file. Subsequently, select "Numerical".
- Fill out the "Fixed effect variable 3" field with "PC2", which is the 13th column label of the "Maize _pheno.csv" file. Subsequently, select "Numerical".
- Fill out the "Fixed effect variable 4" field with "PC3", which is the 14th column label of the "Maize _pheno.csv" file. Subsequently, select "Numerical".
- Click the "Browse" button for "Genotype file", and choose the following file: "Maize _geno.csv".
- Click the "Upload & Submit" button.
- Once the analysis is completed, you can download P value result file. If you want to have GWASpro generating a Manhattan plot, click "manhattan plot" link, which will direct you to "MPlot – Manhattan Plot Draw" page.
- Click the button next to "Marker Coordinates file" field, and choose the "SNP_map_maize.csv" file.
- GWASpro will retrieve a Manhattan plot.
Example - Uploading of input files (Maize data)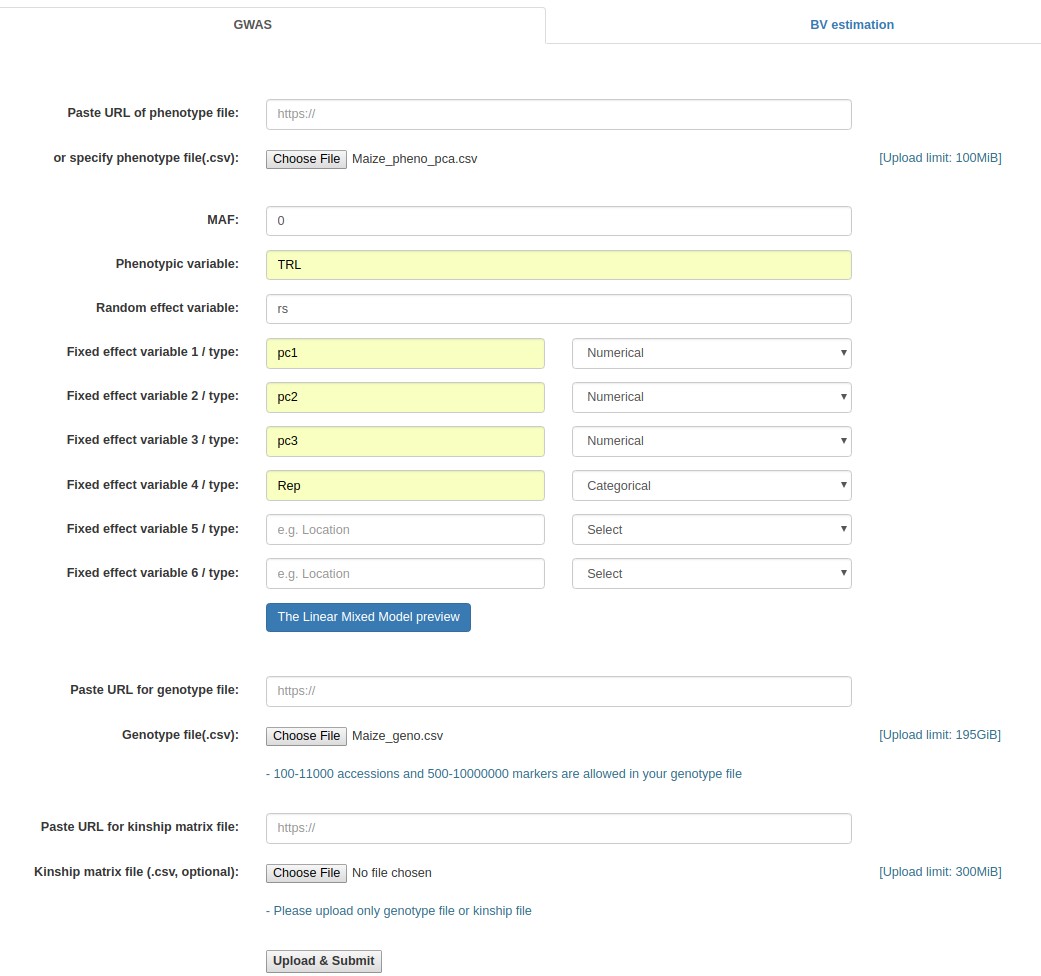
|
|---|
Case study 3: Maize data (Specifying the online paths of the input files)
- Fill out the "Paste URL of phenotype file" field with "http://www.zhaolab.org/GWASPRO/static/demo/Maize_pheno_pca.csv".
- Fill out the "MAF" field with 0.
- Fill out the "Phenotypic variable" field with either "LeafSize" or "shootDryWeight". These are column labels of traits in the "Medicago _pheno.csv" file.
- Fill out the "Random effect variable" field with "id", which is the 1st column label of the "Medicago _pheno.csv" file.
- Fill out the "Fixed effect variable 1" field with "Q1", which is the 5th column label of the "Medicago _pheno.csv" file. Subsequently, select "Numerical".
- Fill out the "Paste URL for genotype file" field with "http://www.zhaolab.org/GWASPRO/static/demo/Maize_geno.csv".
- Click the "Upload & Submit" button.
- Once the analysis is completed, you can download P value result file. If you want to have GWASpro generating a Manhattan plot, click "manhattan plot" link, which will direct you to "MPlot – Manhattan Plot Draw" page.
- Click the button next to "Marker Coordinates file" field, and choose the "SNP_map_medicago.csv" file.
Example - Specifying the online file paths (Maize data)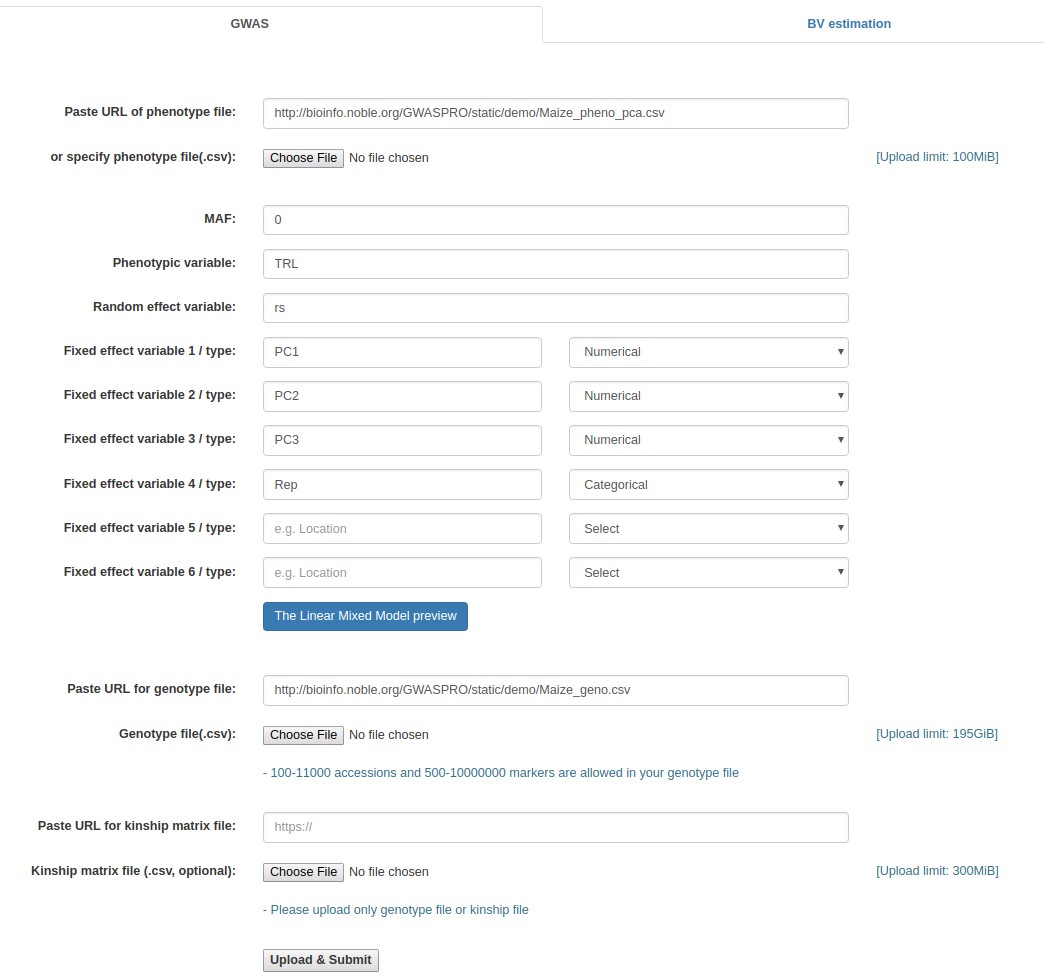
|
|---|
Case study 4: Rice data for estimating breeding values (Uploarding of input files)
- Download the following three files:
- Rice phenotypic data (pheHybrid1500.csv)
- Kinship matrix (kk.csv)
- Click the "Browse" button next to the "Phenotype file" field, go to the folder where the data files are saved, and choose the following file: "pheHybrid1500.csv".
- Fill out the "Phenotypic variable" field with either "KGW".
- Fill out the "Random effect variable" field with "hybrid".
- Fill out the "Fixed effect variable 1" field with "Location".
- Click the "Upload & Submit" button.
- Once the analysis is completed, you can download a resulting file including breeding values.
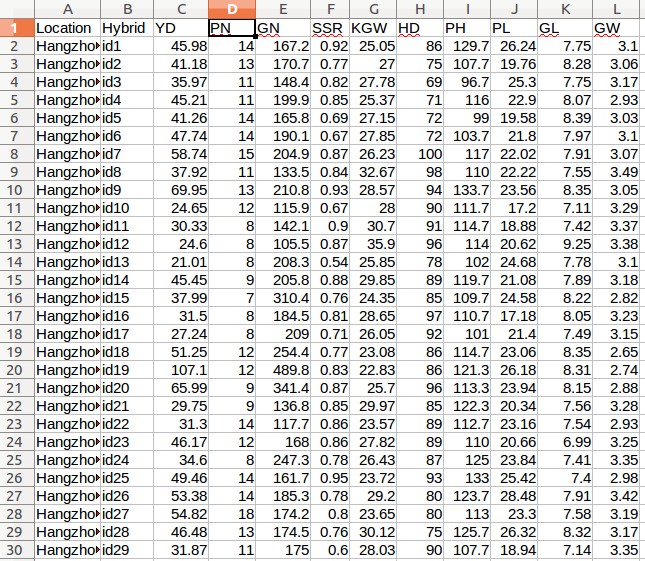
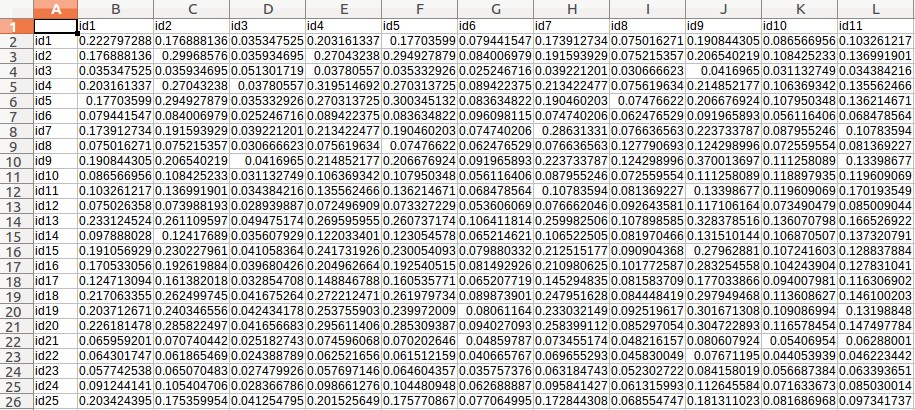
Example - Uploading of input files (Rice data for estimating breeding values)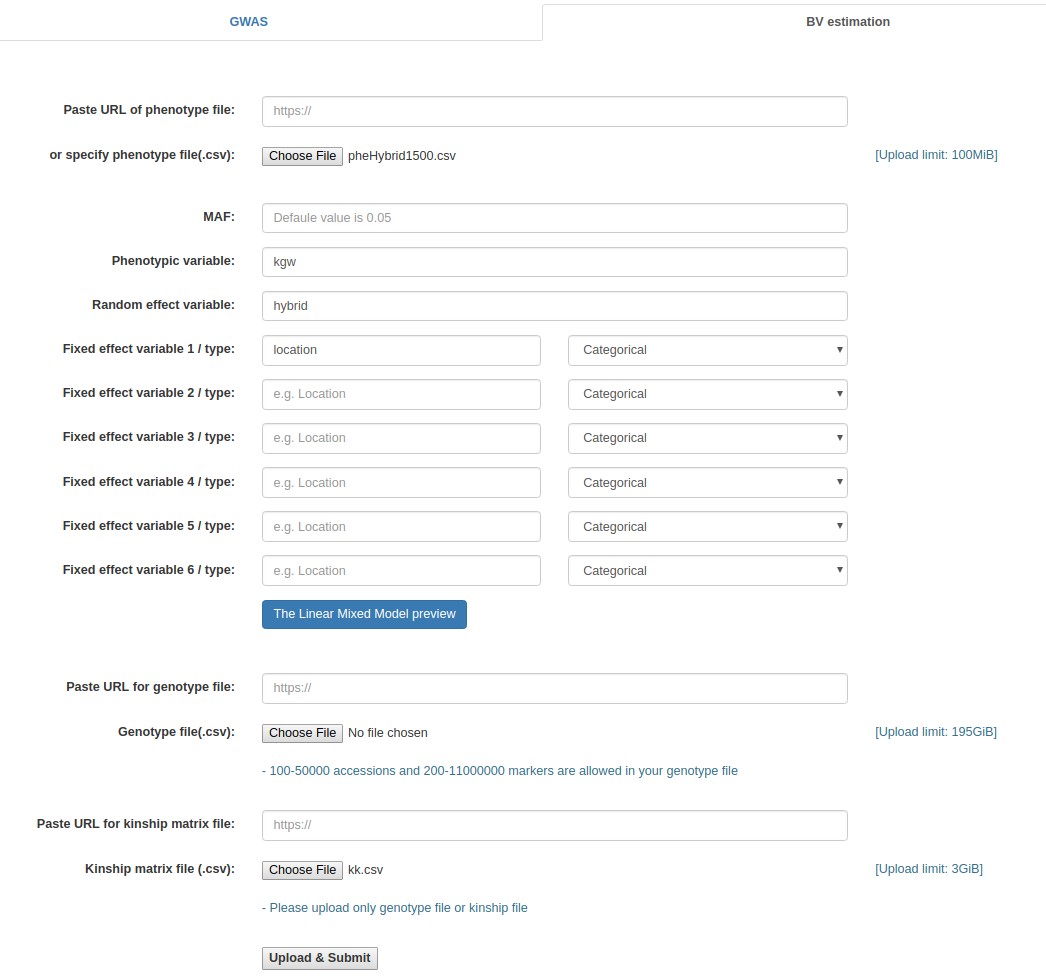
|
|---|
Case study 4: Rice data for estimating breeding values (Specifying the online paths of the input files)
- Fill out the "Paste URL of phenotype file" field with "http://www.zhaolab.org/GWASPRO/static/demo/pheHybrid1500.csv".
- Fill out the "Phenotypic variable" field with either "KGW".
- Fill out the "Random effect variable" field with "hybrid".
- Fill out the "Fixed effect variable 1" field with "Location".
- Fill out the "Paste URL for kinship matrix file" field with "http://www.zhaolab.org/GWASPRO/static/demo/kk.csv".
- Click the "Upload & Submit" button.
- Once the analysis is completed, you can download a resulting file including breeding values.
Example - Specifying the online file paths (Rice data for estimating breeding values)
|
|---|