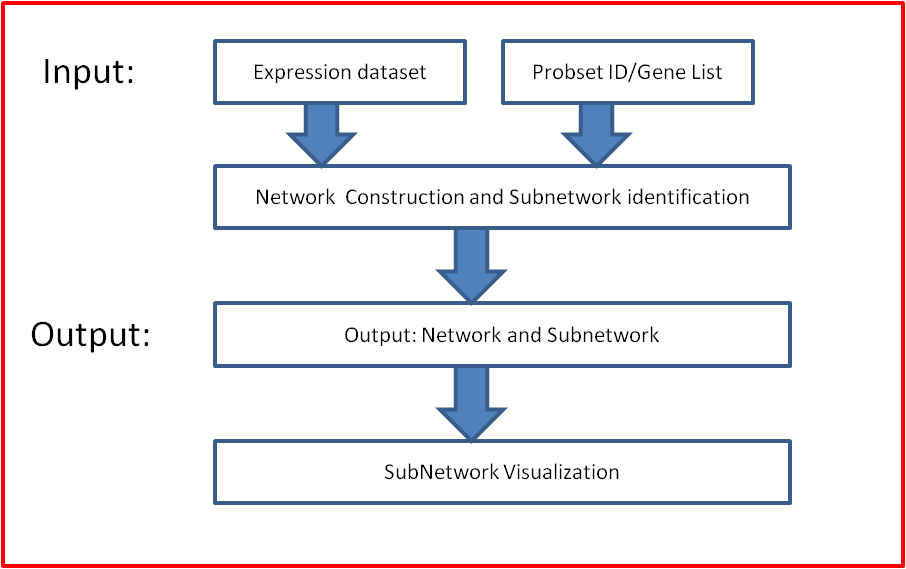
8. Output and Visualization
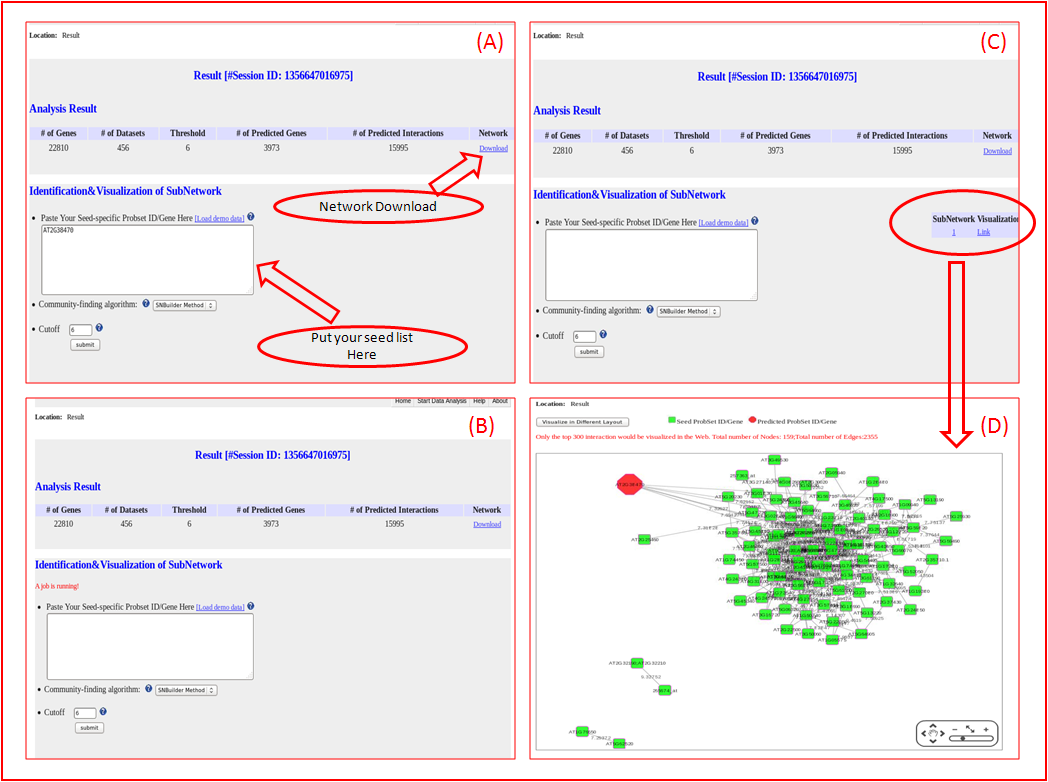
Our web server provided constructed network and subnetwork result for users donwloading. The network result is used to depict the genome-scale gene interactions;
The subnetwork results is used to depict the identified subnetwork based on the constructed network and seeded Probset ID/Gene List.
Meanwhile, our server provide function to visualize this subnetwork based on the Build-in Cytoscape Web Plugin.
7. Reference:
-
1. Faith, J.J., Hayete, B., Thaden, J.T., Mogno, I., Wierzbowski, J., Cottarel, G., Kasif, S., Collins, J.J. and Gardner, T.S. (2007) Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol, 5, e8.
-
2. Kumari, S., Nie, J., Chen, H.S., Ma, H., Stewart, R., Li, X., Lu, M.Z., Taylor, W.M. and Wei, H. (2012) Evaluation of gene association methods for coexpression network construction and biological knowledge discovery. PLoS One, 7, e50411.
-
3. Reshef, D.N., Reshef, Y.A., Finucane, H.K., Grossman, S.R., McVean, G., Turnbaugh, P.J., Lander, E.S., Mitzenmacher, M. and Sabeti, P.C. (2011) Detecting novel associations in large data sets. Science, 334, 1518-1524.
-
4. Aluru, M., Zola, J., Nettleton, D. and Aluru, S. (2012) Reverse engineering and analysis of large genome-scale gene networks. Nucleic Acids Res.
-
5. Hu, X. and Wu, F.X. (2007) Mining and state-space modeling and verification of sub-networks from large-scale biomolecular networks. BMC Bioinformatics, 8, 324.
- 6. Kohl, M., Wiese, S. and Warscheid, B. (2011) Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol, 696, 291-303.
-
7. Parkinson, H., Kapushesky, M., Shojatalab, M., Abeygunawardena, N., Coulson, R., Farne, A., Holloway, E., Kolesnykov, N., Lilja, P., Lukk, M. et al. (2007) ArrayExpress--a public database of microarray experiments and gene expression profiles. Nucleic Acids Res, 35, D747-750.
- 8. Qiu, J.L., Fiil, B.K., Petersen, K., Nielsen, H.B., Botanga, C.J., Thorgrimsen, S., Palma, K., Suarez-Rodriguez, M.C., Sandbech-Clausen, S., Lichota, J. et al. (2008) Arabidopsis MAP kinase 4 regulates gene expression through transcription factor release in the nucleus. EMBO J, 27, 2214-2221.
- 9. Birkenbihl, R.P., Diezel, C. and Somssich, I.E. (2012) Arabidopsis WRKY33 is a key transcriptional regulator of hormonal and metabolic responses toward Botrytis cinerea infection. Plant Physiol, 159, 266-285.
8. Funding:
- This work was supported by National Science Foundation (Grant ABI-0960897) and the Samuel Roberts Noble Foundation.
9. Contact:
- To contact us, please write to: bioinfo AT noble DOT org
|